

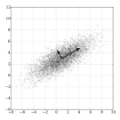
With the ongoing COVID-19 pandemic, understanding the characteristics of the virus has become an important and challenging task in the scientific community. While tests do exist for COVID-19, the goal of our research is to explore other methods of identifying infected individuals. Our group applied unsupervised clustering techniques to explore a dataset of lungscans of COVID-19 infected, Viral Pneumonia infected, and healthy individuals. This is an important area to explore as COVID-19 is a novel disease that is currently being studied in detail. Our methodology explores the potential that unsupervised clustering algorithms have to reveal important hidden differences between COVID-19 and other respiratory illnesses. Our experiments use: Principal Component Analysis (PCA), K-Means++ (KM++) and the recently developed Robust Continuous Clustering algorithm (RCC). We evaluate the performance of KM++ and RCC in clustering COVID-19 lung scans using the Adjusted Mutual Information (AMI) score.
相關內容
Clustering points in a vector space or nodes in a graph is a ubiquitous primitive in statistical data analysis, and it is commonly used for exploratory data analysis. In practice, it is often of interest to "refine" or "improve" a given cluster that has been obtained by some other method. In this survey, we focus on principled algorithms for this cluster improvement problem. Many such cluster improvement algorithms are flow-based methods, by which we mean that operationally they require the solution of a sequence of maximum flow problems on a (typically implicitly) modified data graph. These cluster improvement algorithms are powerful, both in theory and in practice, but they have not been widely adopted for problems such as community detection, local graph clustering, semi-supervised learning, etc. Possible reasons for this are: the steep learning curve for these algorithms; the lack of efficient and easy to use software; and the lack of detailed numerical experiments on real-world data that demonstrate their usefulness. Our objective here is to address these issues. To do so, we guide the reader through the whole process of understanding how to implement and apply these powerful algorithms. We present a unifying fractional programming optimization framework that permits us to distill, in a simple way, the crucial components of all these algorithms. It also makes apparent similarities and differences between related methods. Viewing these cluster improvement algorithms via a fractional programming framework suggests directions for future algorithm development. Finally, we develop efficient implementations of these algorithms in our LocalGraphClustering Python package, and we perform extensive numerical experiments to demonstrate the performance of these methods on social networks and image-based data graphs.
The recent development of scintillation crystals combined with $\gamma$-rays sources opens the way to an imaging concept based on Compton scattering, namely Compton scattering tomography (CST). The associated inverse problem rises many challenges: non-linearity, multiple order-scattering and high level of noise. Already studied in the literature, these challenges lead unavoidably to uncertainty of the forward model. This work proposes to study exact and approximated forward models and develops two data-driven reconstruction algorithms able to tackle the inexactness of the forward model. The first one is based on the projective method called regularized sequential subspace optimization (RESESOP). We consider here a finite dimensional restriction of the semi-discrete forward model and show its well-posedness and regularisation properties. The second one considers the unsupervised learning method, deep image prior (DIP), inspired by the construction of the model uncertainty in RESESOP. The methods are validated on Monte-Carlo data.
The overwhelming presence of categorical/sequential data in diverse domains emphasizes the importance of sequence mining. The challenging nature of sequences proves the need for continuing research to find a more accurate and faster approach providing a better understanding of their (dis)similarities. This paper proposes a new Model-based approach for clustering sequence data, namely nTreeClus. The proposed method deploys Tree-based Learners, k-mers, and autoregressive models for categorical time series, culminating with a novel numerical representation of the categorical sequences. Adopting this new representation, we cluster sequences, considering the inherent patterns in categorical time series. Accordingly, the model showed robustness to its parameter. Under different simulated scenarios, nTreeClus improved the baseline methods for various internal and external cluster validation metrics for up to 10.7% and 2.7%, respectively. The empirical evaluation using synthetic and real datasets, protein sequences, and categorical time series showed that nTreeClus is competitive or superior to most state-of-the-art algorithms.
We propose a general approach for distance based clustering, using the gradient of the cost function that measures clustering quality with respect to cluster assignments and cluster center positions. The approach is an iterative two step procedure (alternating between cluster assignment and cluster center updates) and is applicable to a wide range of functions, satisfying some mild assumptions. The main advantage of the proposed approach is a simple and computationally cheap update rule. Unlike previous methods that specialize to a specific formulation of the clustering problem, our approach is applicable to a wide range of costs, including non-Bregman clustering methods based on the Huber loss. We analyze the convergence of the proposed algorithm, and show that it converges to the set of appropriately defined fixed points, under arbitrary center initialization. In the special case of Bregman cost functions, the algorithm converges to the set of centroidal Voronoi partitions, which is consistent with prior works. Numerical experiments on real data demonstrate the effectiveness of the proposed method.
In semi-supervised domain adaptation, a few labeled samples per class in the target domain guide features of the remaining target samples to aggregate around them. However, the trained model cannot produce a highly discriminative feature representation for the target domain because the training data is dominated by labeled samples from the source domain. This could lead to disconnection between the labeled and unlabeled target samples as well as misalignment between unlabeled target samples and the source domain. In this paper, we propose a novel approach called Cross-domain Adaptive Clustering to address this problem. To achieve both inter-domain and intra-domain adaptation, we first introduce an adversarial adaptive clustering loss to group features of unlabeled target data into clusters and perform cluster-wise feature alignment across the source and target domains. We further apply pseudo labeling to unlabeled samples in the target domain and retain pseudo-labels with high confidence. Pseudo labeling expands the number of ``labeled" samples in each class in the target domain, and thus produces a more robust and powerful cluster core for each class to facilitate adversarial learning. Extensive experiments on benchmark datasets, including DomainNet, Office-Home and Office, demonstrate that our proposed approach achieves the state-of-the-art performance in semi-supervised domain adaptation.
This paper focuses on the expected difference in borrower's repayment when there is a change in the lender's credit decisions. Classical estimators overlook the confounding effects and hence the estimation error can be magnificent. As such, we propose another approach to construct the estimators such that the error can be greatly reduced. The proposed estimators are shown to be unbiased, consistent, and robust through a combination of theoretical analysis and numerical testing. Moreover, we compare the power of estimating the causal quantities between the classical estimators and the proposed estimators. The comparison is tested across a wide range of models, including linear regression models, tree-based models, and neural network-based models, under different simulated datasets that exhibit different levels of causality, different degrees of nonlinearity, and different distributional properties. Most importantly, we apply our approaches to a large observational dataset provided by a global technology firm that operates in both the e-commerce and the lending business. We find that the relative reduction of estimation error is strikingly substantial if the causal effects are accounted for correctly.
The COVID-19 pandemic continues to have a devastating effect on the health and well-being of the global population. A critical step in the fight against COVID-19 is effective screening of infected patients, with one of the key screening approaches being radiological imaging using chest radiography. Motivated by this, a number of artificial intelligence (AI) systems based on deep learning have been proposed and results have been shown to be quite promising in terms of accuracy in detecting patients infected with COVID-19 using chest radiography images. However, to the best of the authors' knowledge, these developed AI systems have been closed source and unavailable to the research community for deeper understanding and extension, and unavailable for public access and use. Therefore, in this study we introduce COVID-Net, a deep convolutional neural network design tailored for the detection of COVID-19 cases from chest radiography images that is open source and available to the general public. We also describe the chest radiography dataset leveraged to train COVID-Net, which we will refer to as COVIDx and is comprised of 5941 posteroanterior chest radiography images across 2839 patient cases from two open access data repositories. Furthermore, we investigate how COVID-Net makes predictions using an explainability method in an attempt to gain deeper insights into critical factors associated with COVID cases, which can aid clinicians in improved screening. By no means a production-ready solution, the hope is that the open access COVID-Net, along with the description on constructing the open source COVIDx dataset, will be leveraged and build upon by both researchers and citizen data scientists alike to accelerate the development of highly accurate yet practical deep learning solutions for detecting COVID-19 cases and accelerate treatment of those who need it the most.
Existing Earth Vision datasets are either suitable for semantic segmentation or object detection. In this work, we introduce the first benchmark dataset for instance segmentation in aerial imagery that combines instance-level object detection and pixel-level segmentation tasks. In comparison to instance segmentation in natural scenes, aerial images present unique challenges e.g., a huge number of instances per image, large object-scale variations and abundant tiny objects. Our large-scale and densely annotated Instance Segmentation in Aerial Images Dataset (iSAID) comes with 655,451 object instances for 15 categories across 2,806 high-resolution images. Such precise per-pixel annotations for each instance ensure accurate localization that is essential for detailed scene analysis. Compared to existing small-scale aerial image based instance segmentation datasets, iSAID contains 15$\times$ the number of object categories and 5$\times$ the number of instances. We benchmark our dataset using two popular instance segmentation approaches for natural images, namely Mask R-CNN and PANet. In our experiments we show that direct application of off-the-shelf Mask R-CNN and PANet on aerial images provide suboptimal instance segmentation results, thus requiring specialized solutions from the research community. The dataset is publicly available at: //captain-whu.github.io/iSAID/index.html
Radiologist is "doctor's doctor", biomedical image segmentation plays a central role in quantitative analysis, clinical diagnosis, and medical intervention. In the light of the fully convolutional networks (FCN) and U-Net, deep convolutional networks (DNNs) have made significant contributions in biomedical image segmentation applications. In this paper, based on U-Net, we propose MDUnet, a multi-scale densely connected U-net for biomedical image segmentation. we propose three different multi-scale dense connections for U shaped architectures encoder, decoder and across them. The highlights of our architecture is directly fuses the neighboring different scale feature maps from both higher layers and lower layers to strengthen feature propagation in current layer. Which can largely improves the information flow encoder, decoder and across them. Multi-scale dense connections, which means containing shorter connections between layers close to the input and output, also makes much deeper U-net possible. We adopt the optimal model based on the experiment and propose a novel Multi-scale Dense U-Net (MDU-Net) architecture with quantization. Which reduce overfitting in MDU-Net for better accuracy. We evaluate our purpose model on the MICCAI 2015 Gland Segmentation dataset (GlaS). The three multi-scale dense connections improve U-net performance by up to 1.8% on test A and 3.5% on test B in the MICCAI Gland dataset. Meanwhile the MDU-net with quantization achieves the superiority over U-Net performance by up to 3% on test A and 4.1% on test B.
The availability of large microarray data has led to a growing interest in biclustering methods in the past decade. Several algorithms have been proposed to identify subsets of genes and conditions according to different similarity measures and under varying constraints. In this paper we focus on the exclusive row biclustering problem for gene expression data sets, in which each row can only be a member of a single bicluster while columns can participate in multiple ones. This type of biclustering may be adequate, for example, for clustering groups of cancer patients where each patient (row) is expected to be carrying only a single type of cancer, while each cancer type is associated with multiple (and possibly overlapping) genes (columns). We present a novel method to identify these exclusive row biclusters through a combination of existing biclustering algorithms and combinatorial auction techniques. We devise an approach for tuning the threshold for our algorithm based on comparison to a null model in the spirit of the Gap statistic approach. We demonstrate our approach on both synthetic and real-world gene expression data and show its power in identifying large span non-overlapping rows sub matrices, while considering their unique nature. The Gap statistic approach succeeds in identifying appropriate thresholds in all our examples.
小貼士
登錄享
相關主題


注冊地址: 北京市海淀區羊坊店路18號2幢3層301-191