
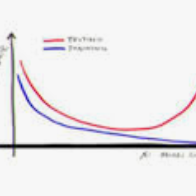

Modern neural networks are often operated in a strongly overparametrized regime: they comprise so many parameters that they can interpolate the training set, even if actual labels are replaced by purely random ones. Despite this, they achieve good prediction error on unseen data: interpolating the training set does not lead to a large generalization error. Further, overparametrization appears to be beneficial in that it simplifies the optimization landscape. Here we study these phenomena in the context of two-layers neural networks in the neural tangent (NT) regime. We consider a simple data model, with isotropic covariates vectors in $d$ dimensions, and $N$ hidden neurons. We assume that both the sample size $n$ and the dimension $d$ are large, and they are polynomially related. Our first main result is a characterization of the eigenstructure of the empirical NT kernel in the overparametrized regime $Nd\gg n$. This characterization implies as a corollary that the minimum eigenvalue of the empirical NT kernel is bounded away from zero as soon as $Nd\gg n$, and therefore the network can exactly interpolate arbitrary labels in the same regime. Our second main result is a characterization of the generalization error of NT ridge regression including, as a special case, min-$\ell_2$ norm interpolation. We prove that, as soon as $Nd\gg n$, the test error is well approximated by the one of kernel ridge regression with respect to the infinite-width kernel. The latter is in turn well approximated by the error of polynomial ridge regression, whereby the regularization parameter is increased by a `self-induced' term related to the high-degree components of the activation function. The polynomial degree depends on the sample size and the dimension (in particular on $\log n/\log d$).
相關內容
We present a novel hybrid algorithm for training Deep Neural Networks that combines the state-of-the-art Gradient Descent (GD) method with a Mixed Integer Linear Programming (MILP) solver, outperforming GD and variants in terms of accuracy, as well as resource and data efficiency for both regression and classification tasks. Our GD+Solver hybrid algorithm, called GDSolver, works as follows: given a DNN $D$ as input, GDSolver invokes GD to partially train $D$ until it gets stuck in a local minima, at which point GDSolver invokes an MILP solver to exhaustively search a region of the loss landscape around the weight assignments of $D$'s final layer parameters with the goal of tunnelling through and escaping the local minima. The process is repeated until desired accuracy is achieved. In our experiments, we find that GDSolver not only scales well to additional data and very large model sizes, but also outperforms all other competing methods in terms of rates of convergence and data efficiency. For regression tasks, GDSolver produced models that, on average, had 31.5% lower MSE in 48% less time, and for classification tasks on MNIST and CIFAR10, GDSolver was able to achieve the highest accuracy over all competing methods, using only 50% of the training data that GD baselines required.
We establish optimal convergence rates up to a log-factor for a class of deep neural networks in a classification setting under a restraint sometimes referred to as the Tsybakov noise condition. We construct classifiers in a general setting where the boundary of the bayes-rule can be approximated well by neural networks. Corresponding rates of convergence are proven with respect to the misclassification error. It is then shown that these rates are optimal in the minimax sense if the boundary satisfies a smoothness condition. Non-optimal convergence rates already exist for this setting. Our main contribution lies in improving existing rates and showing optimality, which was an open problem. Furthermore, we show almost optimal rates under some additional restraints which circumvent the curse of dimensionality. For our analysis we require a condition which gives new insight on the restraint used. In a sense it acts as a requirement for the "correct noise exponent" for a class of functions.
We consider stochastic gradient descent and its averaging variant for binary classification problems in a reproducing kernel Hilbert space. In the traditional analysis using a consistency property of loss functions, it is known that the expected classification error converges more slowly than the expected risk even when assuming a low-noise condition on the conditional label probabilities. Consequently, the resulting rate is sublinear. Therefore, it is important to consider whether much faster convergence of the expected classification error can be achieved. In recent research, an exponential convergence rate for stochastic gradient descent was shown under a strong low-noise condition but provided theoretical analysis was limited to the squared loss function, which is somewhat inadequate for binary classification tasks. In this paper, we show an exponential convergence of the expected classification error in the final phase of the stochastic gradient descent for a wide class of differentiable convex loss functions under similar assumptions. As for the averaged stochastic gradient descent, we show that the same convergence rate holds from the early phase of training. In experiments, we verify our analyses on the $L_2$-regularized logistic regression.
The Neural Tangent Kernel (NTK) has emerged as a powerful tool to provide memorization, optimization and generalization guarantees in deep neural networks. A line of work has studied the NTK spectrum for two-layer and deep networks with at least a layer with $\Omega(N)$ neurons, $N$ being the number of training samples. Furthermore, there is increasing evidence suggesting that deep networks with sub-linear layer widths are powerful memorizers and optimizers, as long as the number of parameters exceeds the number of samples. Thus, a natural open question is whether the NTK is well conditioned in such a challenging sub-linear setup. In this paper, we answer this question in the affirmative. Our key technical contribution is a lower bound on the smallest NTK eigenvalue for deep networks with the minimum possible over-parameterization: the number of parameters is roughly $\Omega(N)$ and, hence, the number of neurons is as little as $\Omega(\sqrt{N})$. To showcase the applicability of our NTK bounds, we provide two results concerning memorization capacity and optimization guarantees for gradient descent training.
While neural networks have been remarkably successful in a wide array of applications, implementing them in resource-constrained hardware remains an area of intense research. By replacing the weights of a neural network with quantized (e.g., 4-bit, or binary) counterparts, massive savings in computation cost, memory, and power consumption are attained. To that end, we generalize a post-training neural-network quantization method, GPFQ, that is based on a greedy path-following mechanism. Among other things, we propose modifications to promote sparsity of the weights, and rigorously analyze the associated error. Additionally, our error analysis expands the results of previous work on GPFQ to handle general quantization alphabets, showing that for quantizing a single-layer network, the relative square error essentially decays linearly in the number of weights -- i.e., level of over-parametrization. Our result holds across a range of input distributions and for both fully-connected and convolutional architectures thereby also extending previous results. To empirically evaluate the method, we quantize several common architectures with few bits per weight, and test them on ImageNet, showing only minor loss of accuracy compared to unquantized models. We also demonstrate that standard modifications, such as bias correction and mixed precision quantization, further improve accuracy.
Deep learning plays a more and more important role in our daily life due to its competitive performance in multiple industrial application domains. As the core of DL-enabled systems, deep neural networks automatically learn knowledge from carefully collected and organized training data to gain the ability to predict the label of unseen data. Similar to the traditional software systems that need to be comprehensively tested, DNNs also need to be carefully evaluated to make sure the quality of the trained model meets the demand. In practice, the de facto standard to assess the quality of DNNs in industry is to check their performance (accuracy) on a collected set of labeled test data. However, preparing such labeled data is often not easy partly because of the huge labeling effort, i.e., data labeling is labor-intensive, especially with the massive new incoming unlabeled data every day. Recent studies show that test selection for DNN is a promising direction that tackles this issue by selecting minimal representative data to label and using these data to assess the model. However, it still requires human effort and cannot be automatic. In this paper, we propose a novel technique, named Aries, that can estimate the performance of DNNs on new unlabeled data using only the information obtained from the original test data. The key insight behind our technique is that the model should have similar prediction accuracy on the data which have similar distances to the decision boundary. We performed a large-scale evaluation of our technique on 13 types of data transformation methods. The results demonstrate the usefulness of our technique that the estimated accuracy by Aries is only 0.03% -- 2.60% (on average 0.61%) off the true accuracy. Besides, Aries also outperforms the state-of-the-art selection-labeling-based methods in most (96 out of 128) cases.
The value of uncertainty quantification on predictions for trained neural networks (NNs) on quantum chemical reference data is quantitatively explored. For this, the architecture of the PhysNet NN was suitably modified and the resulting model was evaluated with different metrics to quantify calibration, quality of predictions, and whether prediction error and the predicted uncertainty can be correlated. The results from training on the QM9 database and evaluating data from the test set within and outside the distribution indicate that error and uncertainty are not linearly related. The results clarify that noise and redundancy complicate property prediction for molecules even in cases for which changes - e.g. double bond migration in two otherwise identical molecules - are small. The model was then applied to a real database of tautomerization reactions. Analysis of the distance between members in feature space combined with other parameters shows that redundant information in the training dataset can lead to large variances and small errors whereas the presence of similar but unspecific information returns large errors but small variances. This was, e.g., observed for nitro-containing aliphatic chains for which predictions were difficult although the training set contained several examples for nitro groups bound to aromatic molecules. This underlines the importance of the composition of the training data and provides chemical insight into how this affects the prediction capabilities of a ML model. Finally, the approach put forward can be used for information-based improvement of chemical databases for target applications through active learning optimization.
Neural Tangent Kernel (NTK) is widely used to analyze overparametrized neural networks due to the famous result by Jacot et al. (2018): in the infinite-width limit, the NTK is deterministic and constant during training. However, this result cannot explain the behavior of deep networks, since it generally does not hold if depth and width tend to infinity simultaneously. In this paper, we study the NTK of fully-connected ReLU networks with depth comparable to width. We prove that the NTK properties depend significantly on the depth-to-width ratio and the distribution of parameters at initialization. In fact, our results indicate the importance of the three phases in the hyperparameter space identified in Poole et al. (2016): ordered, chaotic and the edge of chaos (EOC). We derive exact expressions for the NTK dispersion in the infinite-depth-and-width limit in all three phases and conclude that the NTK variability grows exponentially with depth at the EOC and in the chaotic phase but not in the ordered phase. We also show that the NTK of deep networks may stay constant during training only in the ordered phase and discuss how the structure of the NTK matrix changes during training.
Predictive coding (PC) is an influential theory in computational neuroscience, which argues that the cortex forms unsupervised world models by implementing a hierarchical process of prediction error minimization. PC networks (PCNs) are trained in two phases. First, neural activities are updated to optimize the network's response to external stimuli. Second, synaptic weights are updated to consolidate this change in activity -- an algorithm called \emph{prospective configuration}. While previous work has shown how in various limits, PCNs can be found to approximate backpropagation (BP), recent work has demonstrated that PCNs operating in this standard regime, which does not approximate BP, nevertheless obtain competitive training and generalization performance to BP-trained networks while outperforming them on tasks such as online, few-shot, and continual learning, where brains are known to excel. Despite this promising empirical performance, little is understood theoretically about the properties and dynamics of PCNs in this regime. In this paper, we provide a comprehensive theoretical analysis of the properties of PCNs trained with prospective configuration. We first derive analytical results concerning the inference equilibrium for PCNs and a previously unknown close connection relationship to target propagation (TP). Secondly, we provide a theoretical analysis of learning in PCNs as a variant of generalized expectation-maximization and use that to prove the convergence of PCNs to critical points of the BP loss function, thus showing that deep PCNs can, in theory, achieve the same generalization performance as BP, while maintaining their unique advantages.
Graph convolution networks (GCN) are increasingly popular in many applications, yet remain notoriously hard to train over large graph datasets. They need to compute node representations recursively from their neighbors. Current GCN training algorithms suffer from either high computational costs that grow exponentially with the number of layers, or high memory usage for loading the entire graph and node embeddings. In this paper, we propose a novel efficient layer-wise training framework for GCN (L-GCN), that disentangles feature aggregation and feature transformation during training, hence greatly reducing time and memory complexities. We present theoretical analysis for L-GCN under the graph isomorphism framework, that L-GCN leads to as powerful GCNs as the more costly conventional training algorithm does, under mild conditions. We further propose L^2-GCN, which learns a controller for each layer that can automatically adjust the training epochs per layer in L-GCN. Experiments show that L-GCN is faster than state-of-the-arts by at least an order of magnitude, with a consistent of memory usage not dependent on dataset size, while maintaining comparable prediction performance. With the learned controller, L^2-GCN can further cut the training time in half. Our codes are available at //github.com/Shen-Lab/L2-GCN.
小貼士
登錄享
相關主題




注冊地址: 北京市海淀區羊坊店路18號2幢3層301-191